
がん細胞死を誘導する人工配列の核酸の創製に成功 -核酸医薬の開発に期待-

東京工科大学(東京都八王子市片倉町、学長:軽部征夫)大学院バイオニクス専攻の杉山友康教授らの研究グループは、プログラムされた細胞死(アポトーシス)を、がん細胞に誘導する新しい核酸の創製に成功しました。これは、同研究グループが合成した約15万種類の人工核酸の中から発見されたもので、今後、がん細胞死を誘導する核酸医薬品の開発などが期待されます。
本研究成果は、2017年4月22日に科学誌「Biochemical and Biophysical Research Communications」
に掲載されました(注1)。
【背景】
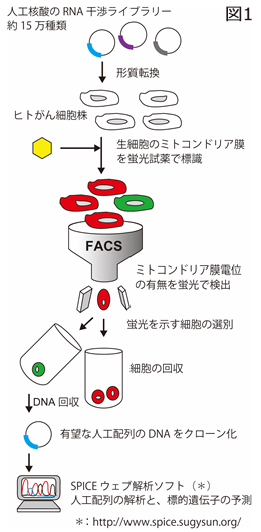
遺伝子の発現を特異的に抑制するRNA(リボ核酸)干渉法は、遺伝子の機能を調べる方法として広く利用されており、近年ではその高い有効性と特異性を活かして医薬品への利用が検討されています。一方で細胞は、個体が恒常性を保つための重要な仕組みとして、自殺(細胞死)するための内在的な機構を持っています。例えば、DNA傷害のような過剰なストレスを受けた場合、自らを消化して存在を抹消する。この内在的な機構には、細胞内のミトコンドリアが関わっており、その膜電位の消失がアポトーシスを誘導すると考えられています。今回の研究では、独自開発した遺伝子情報解析支援システムと人工配列の核酸ライブラリーを利用し、がん細胞のアポトーシスを誘導する核酸の特定を目的としたスクリーニングに取り組みました(図1)
【成果】
本研究では、様々なDNA配列の人工核酸を、ヒト結腸がんの細胞株HCT116に作用させ、ミトコンドリア膜電位の消失を誘導する人工核酸を探索しました。その結果、効果を示す核酸が約15万種類の中から1つ発見。この核酸はヒトゲノム配列と比較して完全一致しない塩基配列であったが、研究グループはその標的遺伝子の特定に成功し、未解明の膜タンパク質「TMEM117」であることを突き止めました(図2)。TMEM117の遺伝子発現抑制は、細胞内の活性酸素種レベルを上げ、ミトコンドリア経路のアポトーシスを誘導し、がん細胞株の増殖性を著しく抑制しました。同様の効果は、ヒト子宮頸がんの細胞株HeLaでも確認されました。またTMEM117の機能として、がん細胞(特に小胞体に過剰なストレスを受けた時)がアポトーシスする反応経路に関わることが示さました。
図1 開発した人工配列の核酸の高効率スクリーニングシステム。RNA干渉用の核酸ライブラリーを導入したがん細胞は、蛍光試薬で細胞標識しました。その効果の有無は高効率、がん細胞の細胞増殖性が著しく抑制されます。
【社会的?学術的なポイント】
研究グループが新たに発見したTMEM117を標的としたRNA干渉は、小胞体ストレスによるミトコンドリア膜電位の消失を伴うがん細胞のアポトーシスを誘導するものです。今後、TMEM117の機能解明が進むことで、がん細胞死を誘導する核酸医薬品の開発が期待されます。
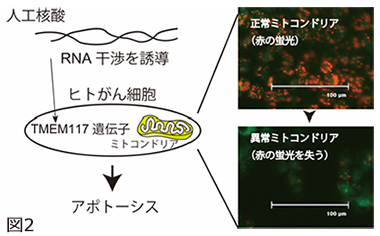
図2 TMEM117を標的とする人工核酸のがん細胞に対する効果。人工核酸はがん細胞内に導入されると、TMEM117の発現を抑制し、がん細胞のミトコンドリア膜電位は消失(図の赤色蛍光が消える)、アポトーシスが誘導されるます。これにより、がん細胞の細胞増殖性が著しく抑制されます。
(注1)オンライン版は2017年3月8日に掲載。論文名「A novel transmembrane protein defines the endoplasmic reticulum stress-induced cell death pathway」
■東京工科大学応用生物学部 杉山友康研究室(機能性RNA工学)
がん細胞の抑制に有効な核酸医薬の開発を目指した研究手法を開発しています。
[主な研究テーマ]
1.がん細胞死?分裂抑制?老化?酸化等を誘導するshRNAの探索とその基礎解析
2.がん幹細胞の分化を誘導するshRNAの探索とその基礎解析
3.六価クロム還元菌のF. alba ST13(T)の応用研究
4.プラナリアをモデル動物とした再生と酸化の基礎解析
【研究内容に関しての報道機関からのお問い合わせ先】
東京工科大学 応用生物学部 教授 杉山友康
Tel 042-637-2477(研究室直通)
E-mail tsugiyama(at)stf.teu.ac.jp
※atはアットマークに置き換えてください。
■応用生物学部WEB
/gakubu/bionics/index.html